Confirming Mutations Created by Genome Editing

Using genome editing to establish mutant strains requires sorting mutations in a variety of processes. However, there remain issues regarding the cost and effort required to confirm the creation of such mutations. This article describes a procedure for the efficient confirmation of created mutations based on the heteroduplex mobility assay [HMA] using the Shimadzu MCE-202 MultiNA microchip electrophoresis system for DNA/RNA analysis.
Genome Editing
Genome editing refers to the genetic modification technology that uses artificial nucleases—designed to break nucleic acid at a particular location in the genome—to create mutations by deleting, inserting, or replacing portions of the genome sequence. Currently, zinc-finger nucleases[ZFNs], transcription activator-like effector nucleases [TAL effector nuclease], and clustered regularly interspaced short palindromic repeats associated protein 9 [CRISPR/Cas9] are the primary artificial nucleases used as genome editing tools. Among these, the use of third-generation CRISPR/Cas9 nucleases is growing rapidly in popularity, due to the ease of designing target sequences.
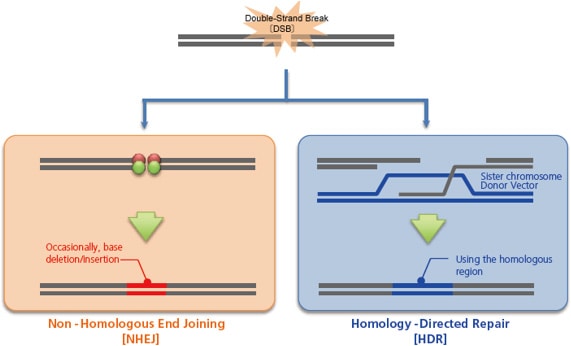
When these artificial nucleases, or vectors that express them, are injected inside a cell by microinjection, electroporation, or other techniques, they break the genome at the target sequence, which the cell subsequently repairs, primarily via one of two pathways. One is non-homologous end joining [NHEJ], which occurs independently of the cell cycle. This pathway tentatively joins the broken genome directly as a stop-gap measure to avoid a genome breakage. However, this process can cause the deletion or insertion of a few bases. This random creation of mutations can be used to intentionally induce a frameshift or gene disruption [knockout].
The other pathway is homology-directed repair [HDR], which uses a normal sister chromosome as a template to guide the repair. This pathway repairs genomes to their original state, and does not result in the types of mutation observed with non-homologous end joining. However, by injecting a homologous double-stranded DNA [or single-stranded DNA] into the cell as a donor, the genome incorporates the sequence from the donor, which enables gene insertion [knock-in] or gene modification.
Genetic modification methods based on genome editing offer dramatically higher modification efficiency than previous methods that depend on natural random recombinations. Furthermore,
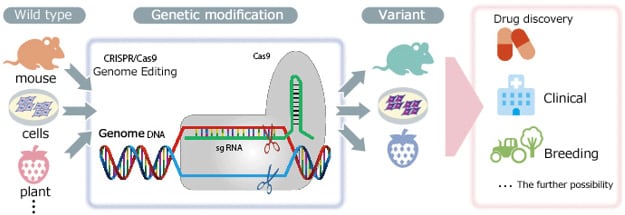
genetic modification was previously only possible in mice and other animals for which embryonic stem cells could be used. Genome editing, however, also enables modification of the genes of other animals, for which creating modifications was hitherto considered difficult, such as for fish, amphibians, reptiles, and large mammals. Genome editing has even been used for genetic modification of insects and plants. Rather than simply remaining at the level of basic research, it is greatly anticipated that genome editing will find use not only in medical fields, such as for creating human genome disease models or gene therapy, but also for developing improved varieties of plants and animals.
Process Flow for Creating Mutations and Mutant Strains
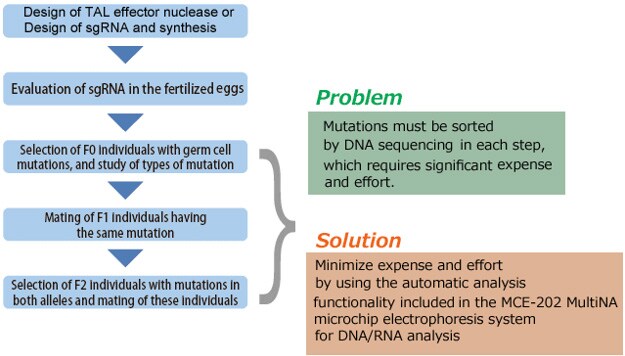
This section describes the process flow for establishing a mutant strain using genome editing. Several steps in the process require considerable expense and effort, such as assessing the activity of TAL effector nuclease or CRISPR/Cas9, and sorting the individuals that harbor the introduced target mutation, the individuals with identical mutations, and the individuals with identical mutations in both alleles. These sorting steps can be accomplished more efficiently by using the HMA process described in the next section.
Confirming a Mutation: Use of the Heteroduplex Mobility Assay [HMA] with the MultiNA System
HMA
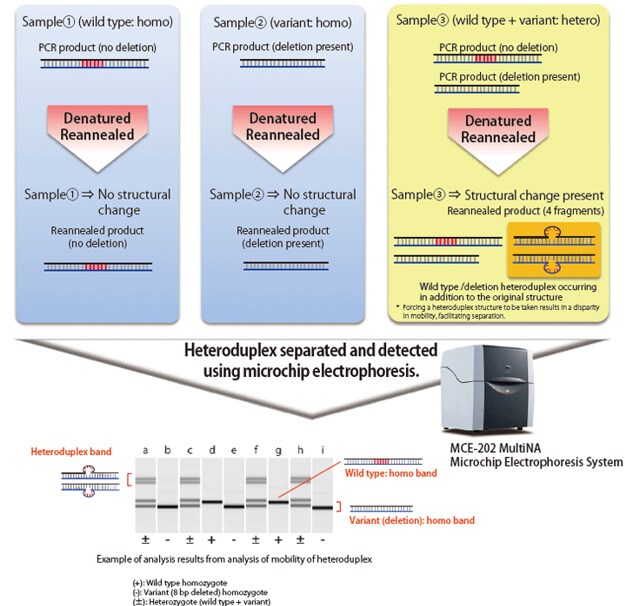
Mutations created by non-homologous end joining for the purpose of knocking out a gene result in small deletions or insertions of one to several dozen bases. Confirming such mutations requires DNA sequencing of the mutated area; however, due to the expense and effort generally required for DNA sequencing, it is more efficient to sort specimens in advance. Due to its convenience and effectiveness, sorting has been accomplished using the HMA, which can be used for sorting based on PCR near the mutation and electrophoresis.
When electrophoresing double-stranded DNA of identical length, the mobility of fully complementary homoduplex DNA depends on its molecular weight. However, with heteroduplex DNA, in which mismatch occurs in some areas, the steric structure of the area containing a mismatch differs from that of homoduplex DNA, which impedes the speed of migration. The HMA process exploits this phenomenon to detect the presence of mutations. Heteroduplex joints are formed by applying PCR near the target sequence in the individual with a created mutation, dissociating the amplification products by heat denaturation, and then reannealing the products. Using electrophoresis, the presence of deletions or insertions can be determined based on electrophoretic patterns.
The MCE-202 MultiNA Microchip Electrophoresis System for DNA/RNA Analysis
⇒ For product details, click here
The MultiNA electrophoresis system is able to automatically analyze up to 108 samples by simply loading the samples and a special reagent into the system. It provides higher separation and detection sensitivity than submarine electrophoresis, but with equivalent running costs. When used for the HMA, the MultiNA system can detect the presence of short deletions and insertions that are difficult to identify using submarine electrophoresis.
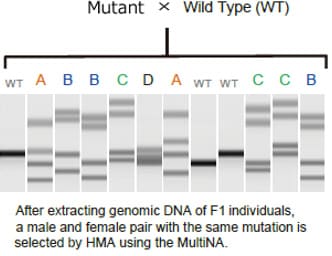
Example of Using the Heteroduplex Mobility Assay to Sort Individuals with Created Mutations
This section describes use of the MultiNA system to obtain F2 individuals by using the HMA to sort out F1 male and female pairs with identical mutations. This involves extracting genomic DNA from a bred F1 individual and then using the MultiNA system to perform the HMA. Multiple band patterns are observed for F1 individuals, as shown in the figure. Excluding wild varieties, there are four types of patterns in the migration example shown, A, B, C, and D. F2 individuals can be obtained by mating sorted male and female F1 individuals that have these sample patterns.
Because F2 individuals are defined as wild, heterozygous mutant, or homo mutant types, in the ratio 1:2:1, the mutant strain can be established by performing the HMA in the same manner and mating sorted male and female homo mutants.
More detail Genome Editing and Creating Mutant Strains in Medaka
Data provided: Faculty/Graduate School of Agriculture, Kyoto University Dr.Masato Kinoshita
The MCE-202 MultiNA Microchip Electrophoresis System for DNA/RNA Analysis
Ideal for Evaluating or Sorting Individuals with Mutations after Genome Editing

Using the MultiNA system to perform the HMA provides an effective way to evaluate and sort mutant individuals. It can differentiate between wild, heterozygous mutant, or homo mutant types based on differences in mobility.
The MultiNA system can accommodate and automatically analyze up to 108 samples. This makes the system particularly useful for mutation screening when a large number of samples must be analyzed.